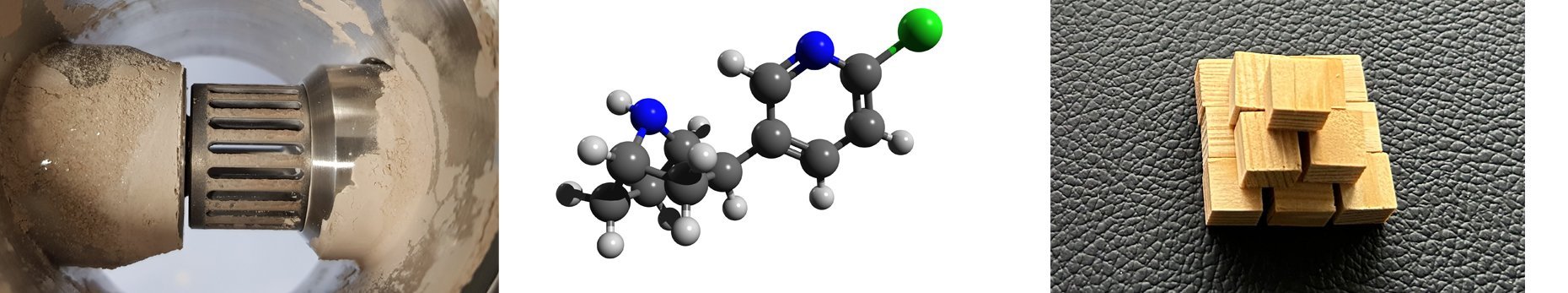
(molecular geometries, boundary orbitals, spectroscopic properties)
The calculation of atomic positions, i.e. distances and bond angles, makes it possible to visualize the complete structure of a molecule. In addition to the geometry of the molecule, the representation of the electronic conditions (HOMO/LUMO calculations and representation of the boundary orbitals) is informative for the chemical reactivity, the exact location of the reaction of a molecule and the way in which the chemical reactions take place.
In the meantime, such quantum chemical calculations, which are usually based on density functional theory (DFT) with suitable combinations of functional and basis set, can be carried out for common non-polymeric organic molecules with powerful, but quite standard computers such as gaming PCs. The software used can run on MS Windows and/or LINUX installations and benefits from the parallel use of as many CPU cores as possible.
The DFT calculations also support the structural determination of new, possibly hypothetical organic substances by calculating infrared molecular spectra (IR), but also electron excitation spectra (UV spectra) and nuclear magnetic resonance spectra (NMR). The latter method is particularly suitable for organic (carbon) compounds, as 13Cand 1H spectroscopydirectly reflect the organic molecular structure.